In klinischen Studien sollen neue Behandlungsverfahren und neue Medikamente getestet werden. Worauf sollten Sie achten, bevor Sie teilnehmen?
Was sind klinische Studien?
Bevor Arzneimittel zugelassen werden, muss ihre Wirksamkeit und Sicherheit in klinischen Studien nachgewiesen werden. Darin wird geprüft werden, ob ein neues Medikament wirksamer ist als ein Scheinpräparat, auch Placebo genannt. Auch neue Behandlungsmethoden werden dahingehend geprüft, ob sie besser sind als der bisherige Behandlungsstandard.
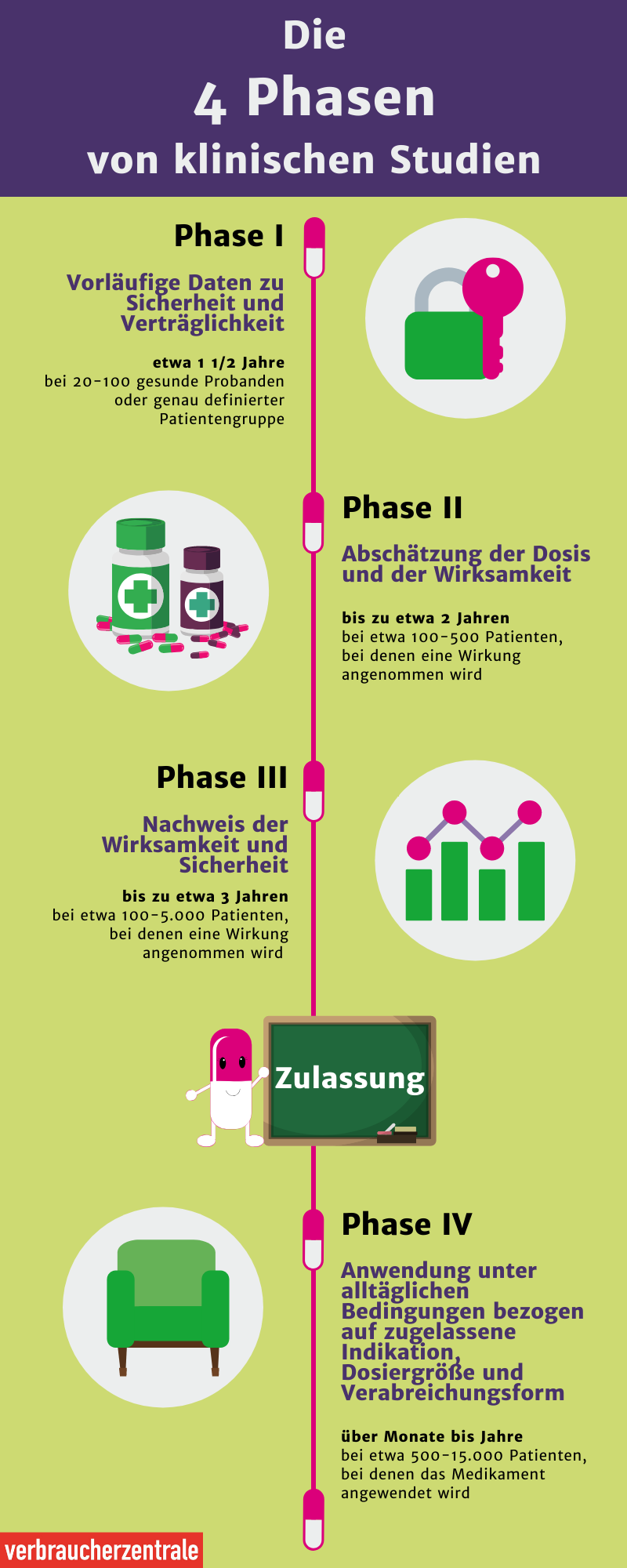
Bevor neue Arzneimittel oder medizinische Behandlungs- und Untersuchungsverfahren am Menschen angewandt werden, werden sie in Tier- sowie Zellversuchen getestet. Waren die Versuche erfolgreich, wird die neue Methode in klinischen Studien am Menschen getestet.
Aus diesem Grund dauerte die Entwicklung eines Impfstoffes gegen Corona eine gewisse Zeit. Im Vergleich zu anderen Impfstoffen wurde der Impfstoff gegen SARS-CoV-2 sehr schnell entwickelt und früh zugelassen. Während die klinischen Phasen üblicherweise mehrere Jahre in Anspruch nehmen, dauerte es hier knapp ein Jahr.
Die Art und Weise, wie eine klinische Studie durchgeführt wird, auch Studiendesign genannt, entscheidet über die Qualität der Ergebnisse.
Höchste Aussagekraft haben Studien, die so durchgeführt wurden:
- placebokontrolliert, in denen der Wirkstoff und ein Scheinmedikament verglichen wurden,
- randomisiert, das heißt, dass die Therapieformen, die man vergleichen will, nach dem Zufallsprinzip zugeordnet werden. Alle Proband:innen werden somit per Zufall entweder der Behandlungsgruppe oder der Kontrollgruppe zugeordnet.
- Doppelblind, das bedeutet, dass weder Arzt noch Proband wissen, welcher Teilnehmer in der Behandlungsgruppe oder der Kontrollgruppe ist, die nur das Placebo bekommt.
Es ist gesetzlich vorgeschrieben, dass eine Ethik-Kommission jeder kontrollierten Studie zustimmen muss, bei der ein neues Medikament oder eine neue Behandlung getestet werden soll. Die Ethik-Kommission muss abwägen, ob der Nutzen der Studie vor möglichen Risiken überwiegt.
Was geschieht mit den Daten aus klinischen Studien?
Wenn die Phase-III-Studien erfolgreich waren, können die Arzneimittelhersteller die Zulassung für das Medikament beantragen. Bevor das Medikament vom Arzt verschrieben werden kann, prüfen die Zulassungsbehörden die Ergebnisse aus den klinischen Studien.
Zulassungsbehörden sind
- das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM)
- das Paul-Ehrlich-Institut für Impfstoffe und biomedizinische Arzneimittel (PEI) und
- in Europa die Europäische Arzneimittelagentur (EMA European Medicines Agency)
Wie bei jeder anderen Behandlung legt der Arzt oder die Ärztin Dokumente an. Darin sind alle Angaben über die Erkrankung, die Behandlungsmaßnahmen und die Untersuchungsergebnisse enthalten. Anders als sonst, muss bei einer Studie ein Teil der Daten weitergegeben werden.
Diese Weitergabe erfolgt verschlüsselt. Nur wenige Institutionen dürfen diese Daten einsehen. Dazu gehören der Auftraggeber, die Überwachungsbehörde sowie die Bundesbehörde, die die Studie genehmigt hat. Bevor Sie an einer Studie teilnehmen, müssen Sie sich damit einverstanden erklären, dass Ihre Daten weitergegeben werden. Die Daten werden auch dann weitergenutzt, wenn Sie die Studie vorzeitig abbrechen.
Für die Aufbewahrung der Daten gelten die aktuellen Bestimmungen des Datenschutzes. In der Regel werden sie mindestens zehn Jahre aufbewahrt. Alle, die an der Studie beteiligt waren, unterliegen der Schweigepflicht, etwa behandelnde Ärzte und Pflegekräfte. Sie dürfen Ihre Daten nicht an unbeteiligte Dritte wiedergeben.
Welche Vor- oder Nachteile hat es, wenn ich an klinischen Studien teilnehme?
Als Teilnehmer:in können Sie sich mit Verfahren behandeln lassen, die bisher noch nicht allgemein zugänglich sind. Sie werden darüber hinaus besonders intensiv ärztlich betreut und überwacht. Je nach Studie bekommen Sie zudem oft eine finanzielle Aufwandsentschädigung, die sich am zeitlichen Aufwand orientiert.
Allerdings können auch bisher nicht bekannte Gefahren auftreten:
- Neue Methoden können eventuell weniger wirksam oder sogar schädlich sein im Vergleich zu bisherigen Verfahren.
- Patient:innen, die der Kontrollgruppe zugeteilt werden, erhalten ein herkömmliches Medikament oder ein Scheinpräparat.
- Außerdem kann die enge ärztliche Überwachung Sie belasten und mit Einschränkungen verbunden sein. Dazu zählen zum Beispiel konkrete Termine oder Dokumentationspflichten.